
What is ACC?
ACC is the abbreviation for Adrenocortical Carcinoma or Adrenal Cancer.
Adrenocortical Carcinoma is a rare disease in which malignant (cancer) cells form in the outer layer of the adrenal gland. There are two adrenal glands in the body. The adrenal glands are small and shaped like a triangle. One adrenal gland sits on top of each kidney. Adrenocortical carcinoma can occur in both adults and children. Treatment for children, however, is different than treatment for adults.
Adrenal glands play an important role since they produce hormones that help regulate your metabolism, immune system, blood pressure, response to stress and other essential functions.
Because of its rarity it is most likely to be discovered during another routine exam like a scanner or physical exam.
What are the symptoms of adrenal cancer?
Adrenal cancers act in one of two ways: they can cause symptoms related to hormone production (see Cushing syndrome below), or they can cause symptoms linked to the tumor size and compression of other organs. The most common symptom reported by patients with adrenocortical cancer is pain in the back or side (called the flank). Unfortunately, this type of pain is common and does not directly suggest a disease of the adrenal cortex. Moreover, you might not be experiencing pain at all.
In adrenocortical cancer, this symptom is usually from the tumor compressing organs, nerves, and other structures around the adrenal gland. Some people describe feeling full with no appetite because of pressure on the stomach and other abdominal organs.
Fewer than 70 out of 100 (70%) of adrenocortical cancers are only located in the adrenal gland at the time of diagnosis. The reason is that the symptoms are pretty common and can be attributed to other diseases before the right root is found.
Cushing syndrome is often the first sign something is wrong with your adrenal glands (when the role of the pituitary gland has been excluded). It is an increased in cortisol or other adrenal gland hormones produced by the adrenal glands.
Symptoms of Cushing syndrome may include:
- Fatty, rounded hump high on the back just below the neck (buffalo hump).
- Flushed, rounded face with pudgy cheeks (moon face).
- Obesity.
- Stunted growth (short stature).
- Virilization – the appearance of male characteristics, including increased body hair (especially on the face), pubic hair, acne, deepening of the voice, and enlarged clitoris (females).
- Mood swings.
- Purple stretch marks especially on the abdomen, thighs, breasts and arms.
- Thinning, fragile skin that bruises easily.
- For women : irregular or absent menstrual periods.
Symptoms of increased aldosterone are the same as symptoms of low potassium, and include:
- Muscle cramps.
- Weakness.
- Pain in the abdomen.
Blood tests will be done to check hormone levels:
- ACTH level will be low.
- Aldosterone level will be high.
- Cortisol level will be high.
- Potassium level will be low.
- Male or female hormones may be abnormally high.
Sources:
“Adrenocortical Carcinoma” by the Cleveland Clinic
“Adrenal Glands” by Johns Hopkins Medicine
“A Patient’s Guide to Adrenocortical Cancer” by Rogel Cancer Center – Michigan Medicine
“Adrenocortical carcinoma” by Mount Sinai
Prognosis
Warning : this part might be hard to read, so if you don’t feel ready please skip and scroll to the next part. As difficult as it might be: facts are facts and we could not avoid talking about them even if our primary focus is on the solution.
We will purposely give a general overview based on our research, but as always please educate yourself and consult your physician or other qualified health provider regarding your health.
Adrenocortical carcinoma is an aggressive cancer with sadly, overall, a poor prognosis. The 5-year survival rate is low, but as you will hear it, the outcome is not predictable and varies a lot.
What is important to know and keep in mind are the factors which help build your prognosis and the treatment you will receive: stage of your cancer, resection status, Ki67 and Weiss Score.
The stage is based on the table established by the European Network for the Study of Adrenal Tumors, which is starting to become the common element of evaluation. It was only put in place in 2009, which tells you how much needs to be done and how difficult it is for ACC patients.
Stage Description
I Tumor < 5 cm, contained in adrenal
II Tumor > 5 cm, contained in adrenal
III Cancer spread to areas near adrenal (vena cava, renal vein)
IV Metastatic disease (the cancer has spread in the body)
Source : ENSAT (European Network for the Study of Adrenal Tumors)
Resection status
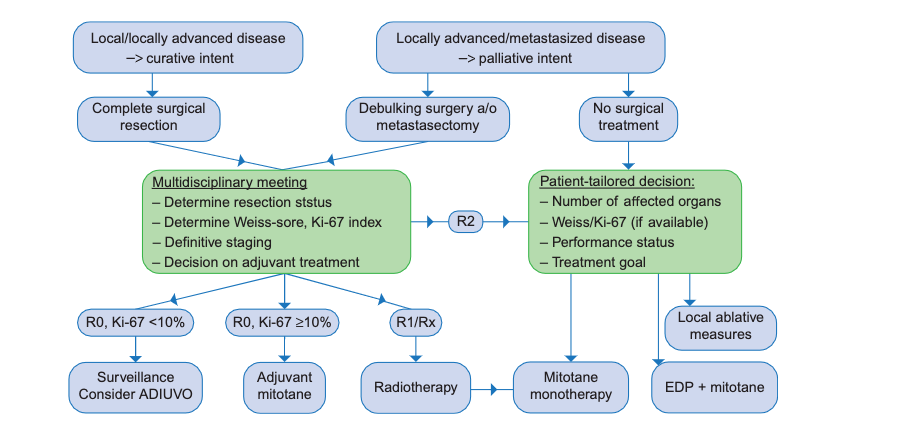
To keep it simple there are 4 statuses. R0, R1, R2 and Rx. The « R » stands for “residual” and the number refers to the degree of resection. It is given after the tumor has been analyzed.
- R0: resection is complete (the whole tumor has been successfully removed) with clear margins.
- R1: resection is incomplete, as some microscopic tumoral tissues cannot be removed.
- R2: resection is incomplete with macroscopic tumoral tissues left (in other words that can be seen by the naked eye).
- Rx: it is impossible to determine the resection status. It means it is unknown.
Ki67
This marker is expressed in percentage. It indicates the chances of proliferation of the tumor, hence the impact on the prognosis. Above 5% means malignancy (aka cancerous). Some papers state that a high level of Ki-67 seems to indicate a worse prognosis with lesser survival rate.
Weiss score
There are 9 criteria and the presence at least of 3 of them indicates malignant potential. This is a very complex data that can be calculated once the tumor is removed by the surgical pathology. It is not easy data to obtain, since the frontier can be blurry in some cases and its lack of objectivity has pointed out by doctors and researchers. Even though some medical experts propose an evolution of the Weiss score, it is an important way to know the gravity of your cancer and it should be included in your follow-up medical report (if possible).
There are not many options available in terms of treatments. We would like to emphasize a point. The course of action will depend on the medical care in your country and what is available to you, but here we will state and sum up the options available in general.
Surgery
Surgery is the best option and the most recommended one since it plays a major role in the prognosis and can be an improving factor. This is true when it comes to the removal of the primary tumor (and its metastases if any occur) as well as for local or distant recurrences. It should be handled by an expert surgeon.
But what happens next? Depending on your situation, you might follow a chemotherapy protocol and/or oral chemotherapy.
Oral chemotherapy / Drug therapy
Oral chemotherapy means taking the only drug available on the market called Lysodren/ Mitotane. It is called an adjuvant therapy since the aim is to decrease the chance of recurrence. It is recommended for cancer with high recurrence risk as well as in advanced disease.
You will need to take as much as you can tolerate, with the aim of reaching the therapeutic level of between 14 mg/L and 20 mg/L. It is said that the drug is most effective between those numbers, with limited side effects. As Mitotane builds up in your body over time, it might take several months to achieve this goal.
2 sides notes:
- Side effects can vary from person to person. If a person is above 20 mg/L, he/she can feel well. Same thing if you have a low Mitotane level.
- Some people will never reach the therapeutic range whatever the number of tablets they take. It does not mean that the drug won’t be effective. A small dose of Mitotane is better than no Mitotane at all.
Chemotherapy
Chemotherapy is used to kill cancer. It can take many forms but in the case of ACC, the product is injected directly in the body (intravenously).
Chemotherapy is the recommended treatment for advanced stages of ACC or recurrence. It is widely accepted that the regimen of EDP (etoposide, doxorubicin, cisplatin) often combined with Mitotane gives the best response rate and is now the “standard of care”.
Based on trials, a combined regimen of gemcitabine/capecitabine is now offered as second-line therapy, especially in Dutch and German expert centers.
Radiotherapy
This is a more localized approach. Only a specific zone is targeted, unlike in chemotherapy where the treatment is injected into the body. The aim is to use radiation to eradicate cancer cells. Just like chemotherapy, radiation therapy affects not only cancer cells, but also healthy cells.
After incomplete surgical resection, radiotherapy in combination with Mitotane is recommended. The rationale behind this recommendation is that patients with residual disease after surgery perform worse compared to patients who had a complete resection.
Radiotherapy with palliative intent, for example irradiation of painful bone metastases or a large irresectable primary tumor, is widely accepted as an effective measure. Evidence is retrospective and mostly based on single-institution experience.
Targeted therapy
Several targeted therapies have been tested, most of them were clinical trials. Unfortunately, at the moment, none of these has yet given proven results as a possible way to treat ACC effectively.
Sources:
“Developing treatment for adrenocortical carcinoma”
“Current and Emerging Therapies for Adrenocortical Carcinoma – Review”
“Role of Mitotane in Adrenocortical Carcinoma – Review and State of the art”